
Continuously Evolving Our NMS Portfolio:
Proven Success, Driven by Ongoing Innovation
Building on our legacy of Oncology expertise, we are advancing innovative
Small Molecule and ADC platforms to drive our strategic focus
|
Program
|
ASSET (Target)
|
PARTNER / LICENSE
|
DISCOVERY
|
IND/
PRE-CLINICAL
|
Phase I
|
Phase II
|
Phase III
|
|
|
Small Molecules
|
Kinases
|
 |
BRAF Mutated Melanoma Combo
BRAF Mutated CRC Combo
BRAF Mutated NSCLC Combo
|
|||||
|
Encorafenib, BRAF inhibitor used in combination for BRAF mutated melanoma, colorectal cancer, non-small cell lung cancer, globally commercialized as BRAFTOVI®. NMS licensed out enabling IP rights, and in 2024, signed a royalty agreement with Blue Owl Capital. | ||||||||


|
ROS Mutated NSCLC
TRK Mutated Solid Tumors
|
|||||||
|
Entrectinib, selective inhibitor of TRK A/B/C, ROS1, and ALK, targeting gene rearrangements that drive tumor growth. Invented by NMS and later licensed to Ignyta, now part of Roche (commercialized under the brand name of ROZLYTREK®). It is approved for patients with NTRK-positive and ROS1-positive metastatic NSCLC, including those with brain metastases, due to its ability to penetrate the blood-brain barrier. March 2025, NMS signed a royalty agreement with NovaQuest Capital Management. | ||||||||
| Exclusive |
Solid Tumors Combo
|
|||||||
|
Onvansertib is the first oral, potent, and selective inhibitor of PLK1, a key regulator of cell division overexpressed in cancer cells. Invented by NMS and licensed to Cardiff Oncology, it is currently in Phase I and II clinical trials. | ||||||||
|
HCC Combo + Atezolizumab
|
||||||||
|
NMS-153 is a potent, selective inhibitor of MPS1/TTK kinase, targeting mitotic cancer cells and inducing cell death through both mitotic catastrophe and immunogenic mechanisms. Currently in Phase I/II clinical trials for hepatocellular carcinoma, it has demonstrated broad in vivo preclinical efficacy as both a single agent and in combination therapies. | ||||||||
|
AML
|
||||||||
|
NMS-812 is a novel, potent dual inhibitor of PERK and GCN2, key effectors of the Integrated Stress Response (ISR) pathway that cancer cells exploit to survive stress. Currently in clinical development with a promising pharmacokinetic profile, it holds potential for overcoming drug resistance in oncology, particularly in Acute Myeloid Leukemia (AML). | ||||||||
|
NAD+ Binding Family: PARP
|

Collaboration / Option to License |
GBM and High-Grade Astrocytoma Combo + TMZ
SCLC Combo + TMZ BRCAwt Ovarian Combo + topotecan
|
||||||
|
NMS-293 is a potent, highly selective PARP1 inhibitor designed to avoid trapping, a known cause of toxicity in healthy cells, making it ideal for combination with DNA-damaging agents like chemotherapies or ADC payloads. Combination dosing is the key for the PARP field to move beyond BRCA mutations. NMS-293 is currently in Phase 2 in relapsed glioblastoma, IDH wild type study , in combination with temozolomide. Initial safety data across the program showed high bone marrow tolerability (Guerts et al, AACR-NCI-EORTC 2023), paving the way for future combinations in other tumors. NMS is developing NMS-293 in GBM, astrocytoma, small cell lung cancer and non-BRCA mutation ovarian cancer. | ||||||||
|
|
||||||||
|
Atamparib is an orally bioavailable, selective small-molecule PARP7 inhibitor designed to inhibit tumor growth in specific cancer types. As a mono-ADP ribosylating enzyme, PARP7 plays a crucial role in controlling key oncogenic pathways that support cancer cell survival. With high selectivity and potency, Atamparib offers a promising approach for addressing high unmet needs in oncology, particularly where PARP7 is a critical factor in disease progression. Atamparib has demonstrated favorable safety across early trials and is poised for a Phase II clinical trial in 2025. | ||||||||
|
ADC Platform
|
Non-Exclusive |
Undisclosed
|
||||||
|
Peptide-Drug Conjugates (PDCs) combine the core benefits of enhanced cellular permeability and improved drug selectivity, enabling targeted delivery to cancer cells. By leveraging the enhanced tumor penetration and tumor-specific binding properties of peptides, PDCs can reduce off-target toxicity and improve therapeutic outcomes. | ||||||||
| Confidential Non-Exclusive |
Undisclosed
|
|||||||
|
Antibody-Drug Conjugates (ADCs) are engineered to deliver highly potent cytotoxic drugs directly to cancer cells via targeted antibodies. This selective approach enhances efficacy while minimizing damage to healthy tissue, making ADCs a powerful tool in precision oncology. | ||||||||
|
Payload linkers are key components of ADCs and PDCs. Selection of the most appropriate payload-linkers, based also on the biology of the target, is essential to ensure the precise release of the drug inside the target cells, optimization of the therapeutic effect and reduced toxicity. | ||||||||
 Encorafenib
Encorafenib
Encorafenib (BRAF)
Encorafenib is a small molecule inhibitor of the BRAF kinase, a key enzyme in the tyrosine kinase/MAPK signaling pathway (TK-RAS-RAF-MEK-ERK), which is often mutated and activated in different tumors, such as melanoma, colorectal cancer, non-small cell lung cancer and others. Encorafenib is a potent BRAF inhibitor for which NMS has licensed enabling IP rights for commercialization. BRAFTOVI® (the brand name for encorafenib) is approved in combination with the MEK inhibitor binimetinib (MEKTOVI®) for the treatment of unresectable or metastatic melanoma with a BRAFV600E or BRAFV600K mutation. It is also the first BRAF inhibitor approved in combination with cetuximab (Erbitux®) for the treatment of adult patients with metastatic colorectal cancer (CRC) with a BRAF V600E mutation. More recently, the FDA and EMA have approved BRAFTOVI® + MEKTOVI® for the treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) with a BRAF V600E mutation. A royalty agreement with Blue Owl Capital has been announced in September 2024. Press release
Entrectinib (TRK, ROS)
Entrectinib is a selective inhibitor of the tyrosine kinases TRK A/B/C, ROS1 and ALK, whose activating gene rearrangements drive proliferation in small subsets of different tumor types. Entrectinib was invented and developed into Phase I by NMS, then licensed to Ignyta (San Diego, USA), a company subsequently acquired by Roche. Entrectinib is one of the first drugs approved with an agnostic indication for adult and pediatric patients with relapsed/refractory tumors harboring NTRK gene fusions, independently from the tumor type. It is also approved for the treatment of adults with ROS1-positive, metastatic non-small cell lung cancer (NSCLC). Entrectinib has been specifically designed to efficiently penetrate the blood brain barrier and it is efficacious also in patients with brain metastases, with a good tolerability profile. A royalty agreement with NovaQuest Capital Management has been announced on March 2025. Press release
Onvansertib (PLK1)
Onvansertib is the first orally available, potent and selective inhibitor of the PLK1 kinase, a master regulator of mitotic progression which is overexpressed and activated in proliferating cancer cells. The drug was invented and developed into Phase I by NMS, then licensed to the US Biotech Trovagene/Cardiff Oncology. Clinical development is currently ongoing with promising results in Phase I/II studies in combination, including a Phase Ib/II study with FOLFIRI/bevacizumab for treatment of metastatic KRAS mut CRC. For a comprehensive, up-to-date, Onvansertib overview please refer to the Cardiff Oncology website.
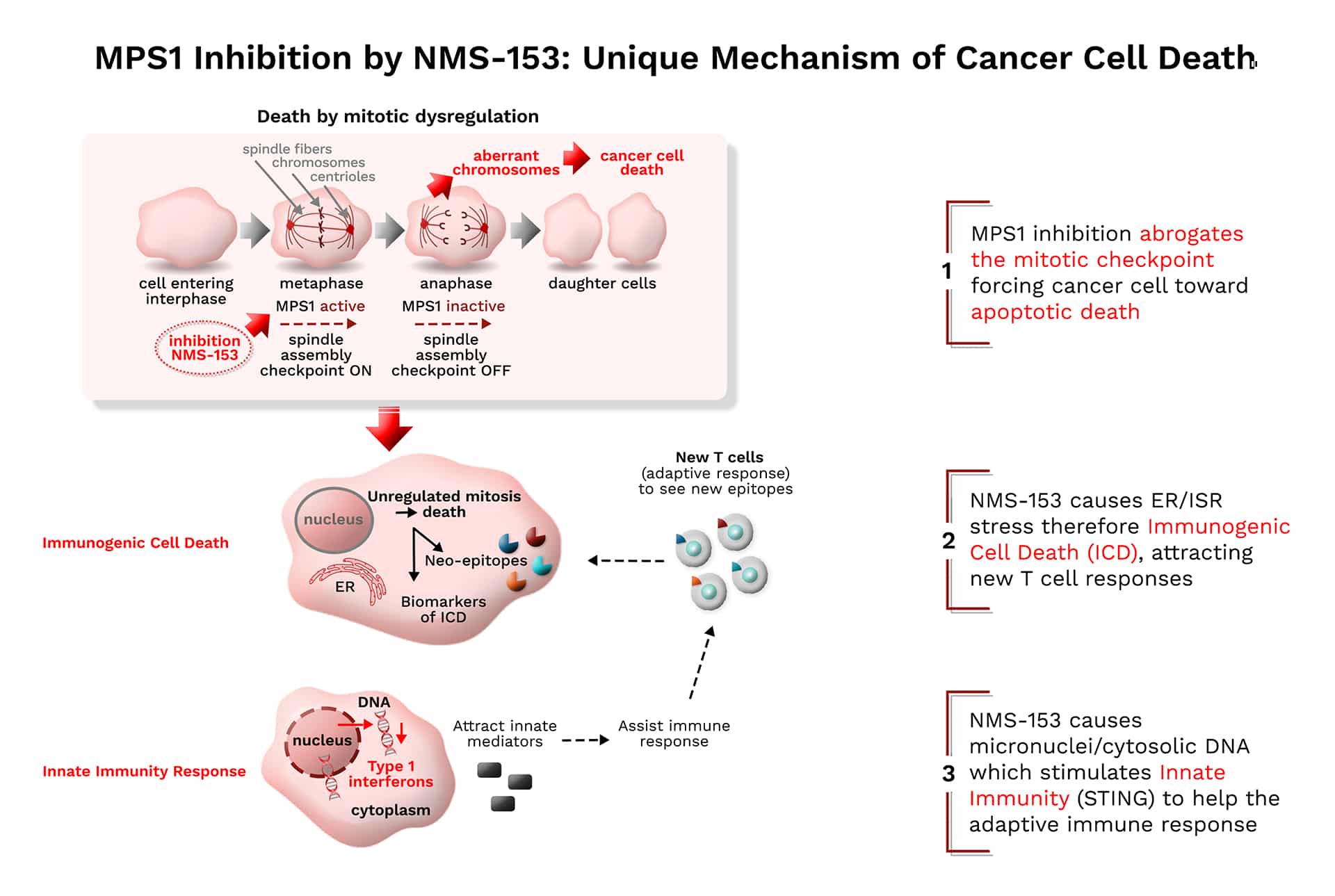
NMS-153 (MPS1)
NMS-153 is a potent and highly selective small molecule inhibitor of Monopolar Spindle 1 (MPS1, also known as TTK kinase) with differentiated, dual anti-cancer mechanism of action. MPS1 is found to be highly expressed in a number of human tumors that play a critical role in the control of mitosis. The mechanism of action of NMS-153 relies upon targeting mitotic cancer cells, causing premature entry into anaphase, before chromosomes are properly aligned at the metaphase plate. This leads to mitotic catastrophe and apoptosis while potentially also harnessing a patient’s immune system by activation of the cGAS/STING pathway and inducing immunogenic cell death.
NMS-153 is a molecule with long residence time on the target and selective against a wide range of tested enzymes. In vitro, a brief exposure to the compound is sufficient to commit cancer cells to death. Its anti-proliferative activity is associated with a demonstrated mechanism of action in different tumor cell lines, including hepatocellular carcinoma and broad in vivo efficacy both as single agent and in combination with standards of care. NMS-153 is currently in Phase II clinical development in hepatocellular carcinoma in combination with atezolizumab (see Press Release).
NMS-812 (PERK/GCN2)
NMS-812 is a novel, potent and orally bioavailable dual inhibitor of PERK (PKR-like endoplasmic reticulum kinase) and GCN2 (General Control Nonderepressible 2), with potential for first-in-class in several potential oncology indications. PERK and GCN2 are effectors of the Integrated Stress Response (ISR), a pro-survival pathway exploited by cancer cells to survive stress. The modulation of ISR may potentially overcome drug resistance and offer superior anti-tumoral activity. In addition, NMS-812 also modulates the immune response via direct and indirect mechanisms which may contribute to anti-cancer activity. The first-in-human (FIH) study showed an excellent pharmacokinetic profile allowing daily oral dosing and likely permissive safety for further development. Based on preclinical data and its unique, dual targeting of the two key components of the Integrated Stress Response PERK and GCN2, NMS-812 may represent a novel strategy for Acute Myeloid Leukemia (AML), with potential for synergies with other drugs and overcoming drug resistance.
NMS-293 (PARP1)
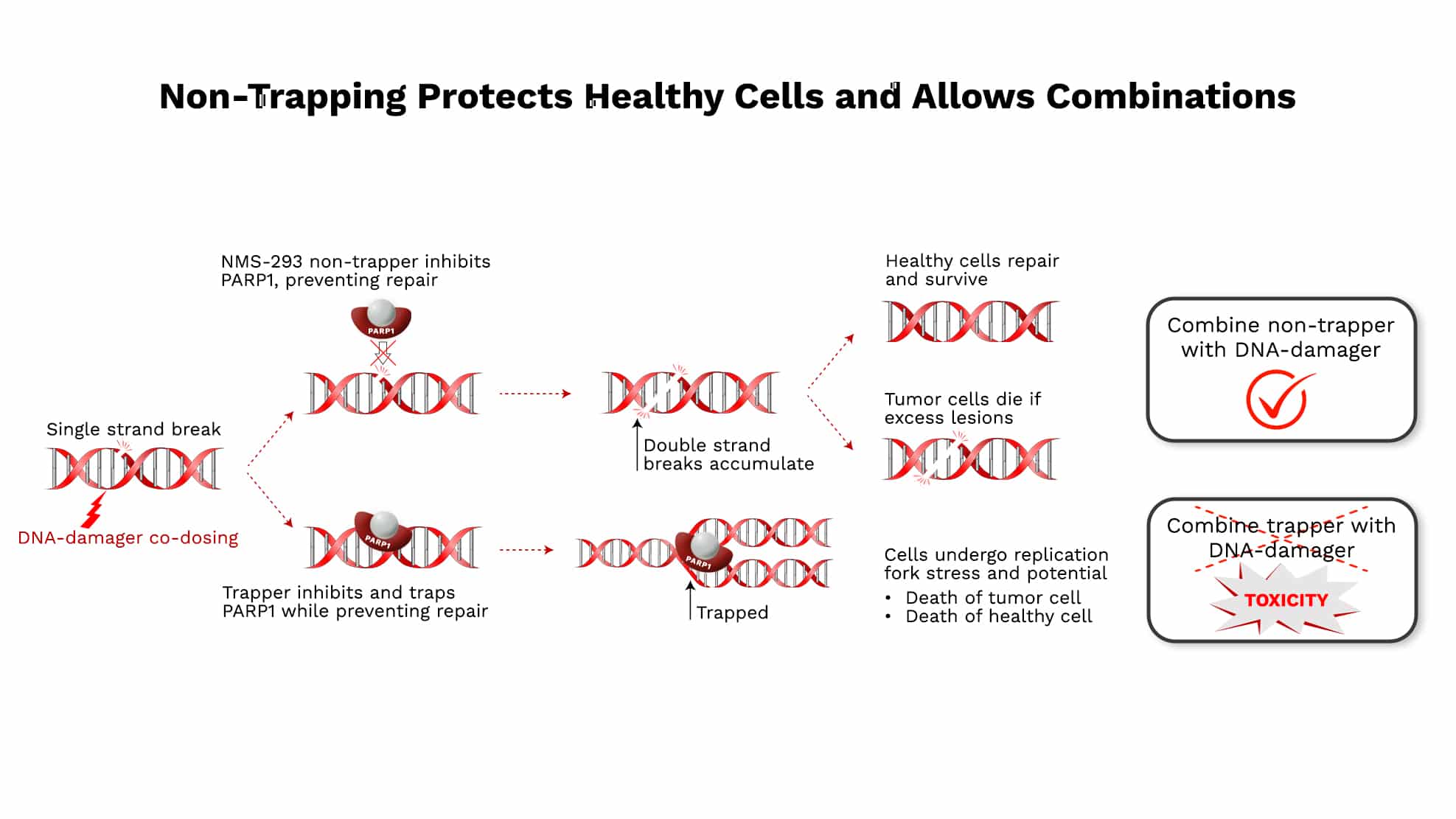
NMS-293 is a unique, intrinsically targeted PARP1 inhibitor. It is the only clinical stage inhibitor that is PARP1-selective with high potency and non-trapping. Recent, emerging, second generation PARP1-selective trapping inhibitors may have similar bone marrow effects relative to marketed PARP1/2 trapping inhibitors; for example: 11% grade 3 neutropenia at the relevant dose of saruparib (Yap et al AACR 2024) being similar to 9-10% for olaparib (Lynparza USPI)
Since trapping is known to cause toxicity in healthy cells (Hopkins et al MCR 2019, Morice et al Lancet Hem 2021), it is possible that trapping PARP inhibitors will continue to have difficulty combining with DNA-damaging drugs or chemotherapies, as has been the case historically. This limitation of combinations will likely hinder development of trapping PARP inhibitors in therapies beyond BRCA mutations or other types of homologous recombination deficiency. Due to non-trapping, many combinations of DNA-damaging agents such as chemotherapies and ADC payloads, have potential for NMS-293 beyond BRCA mutation tumors.
NMS-293 is brain-penetrant, adding value to its non-trapping, PARP1-selective features since it has potential for brain tumors or tumors bearing brain metastases. NMS-293 has completed Phase 1 in a multi-tumor monotherapy study for dose finding (PARPA-293-001, NCT04182516) and is currently in Phase 2 in a high grade glioma study including glioblastoma (GBM) co-dosing with temozolomide (PARPA-293-002, NCT04910022).
Safety findings for NMS-293 are unprecedented for a PARP inhibitor with minimal or negligible bone marrow effects and no exposure relationship of bone marrow parameters, with or without chemotherapy as presented in 2023 (Guerts et al, AACR-NCI-EORTC 2023) and 2024 (Mahnke, DDR Summit). Preliminary trends of confirmed anti-tumor responses are encouraging in combination dosing (Mahnke, DDR Summit).
To maximize the full potential of NMS-293, NMS initiated two Phase 1 clinical trials for the relapse setting: one for BRCA wild-type ovarian cancer (PARPA-293-003 NCT06930755) and another for small cell lung cancer (PARPA-293-004 NCT06931626).
Atamparib (PARP7)
Atamparib is the first-ever developed PARP7 inhibitor, originally created by Ribon Therapeutics and acquired by NMS in November 2024. It enriches NMS’s diverse portfolio with a small molecule targeting PARP7, a critical oncology target upregulated in response to cellular stress, which promotes cancer cell survival. By inhibiting PARP7, atamparib has demonstrated the ability to suppress tumor cell proliferation.
This orally bioavailable and highly selective small molecule has shown high potency in preclinical models and exhibited a favorable safety and tolerability profile and sign of activity in Phase I/II studies (NCT04053673, NCT05127590, JRCT2031210373). Building on Ribon Therapeutics’ foundational research, recent advancements in PARP7 biology and our extensive knowledge in the biology of PARP family, NMS plans to initiate proof-of-concept clinical trials in 2025. These trials will explore diverse cancer settings, reinforcing NMS’s leadership in developing PARP inhibitors.